
UCSD Scientists Use SDSC Supercomputers to Speed Drug Discovery
Published April 13, 2005
More than two decades after it burst onto the scene, HIV/AIDS has claimed more than twenty million lives and continues to devastate societies around the world, particularly in Africa and other developing countries. In the late 1980s and early 1990s, after years of effort AIDS researchers succeeded in developing a class of drugs that proved to be highly effective against AIDS. By blocking the activity of the viral enzyme HIV protease, these protease inhibitors brought greatly extended life-spans to patients who previously faced early deaths. Structural biology and molecular dynamics computer simulations played an important role in these discoveries.
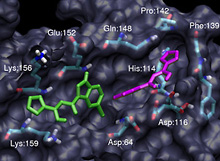
A New Drug Target: HIV Integrase Two different spatial arrangements for inhibitor binding to the HIV-1 integrase enzyme. The green structure on the left is similar to that found by traditional static x-ray crystallography methods. The purple structure on the right is in the novel binding orientation discovered by using computer simulations to "put the molecules in motion" and reflects the unexpectedly large structural fluctuations found in the active site. The large-scale simulations were run by the McCammon group using SDSC supercomputers. McCammon group, UCSD. |
"Fortunately, in this ongoing battle, computational and structural biology may once again be on the verge of contributing a new class of drugs for the treatment of HIV/AIDS," said J. Andrew McCammon, a Howard Hughes Medical Institute investigator and UCSD professor of Pharmacology who holds the Joseph Mayer Chair of Theoretical Chemistry at UCSD. "This time, we're targeting HIV integrase, the only HIV enzyme for which no effective drugs have previously been discovered." The other two HIV enzymes are reverse transcriptase, the target for AZT and other drugs, and protease, the target for the protease inhibitors that had proven so effective until the recent emergence of drug-resistant mutants.
The new drug possibilities flow out of a long series of molecular dynamics simulations by McCammon's group. These simulations have focused on the catalytic domain of HIV-1 integrase, and relied on the large-scale computational resources of the San Diego Supercomputer Center (SDSC) at UC San Diego. In 1999, related results from traditional x-ray crystallography studies of the structure of a complex between the HIV-1 integrase catalytic domain and an inhibitor pointed to the existence of only one site where inhibitors could bind. These studies indicated a single format for small inhibitors of integrase to bind at the catalytic site, typically with a cyclic moiety, or "wing," to the "west" of the active center.
Around this same time, the McCammon group began new molecular dynamics studies of HIV-1 integrase using SDSC's supercomputers. As the power of computers and the sophistication of the scientists' simulation methods have advanced hand in hand, the researchers have been able to discern ever more detailed insights into the microscopic dance of molecular dynamics, where events take place in nanoseconds at length scales far shorter than the wavelength of light. By putting the molecules into motion in their simulations, the researchers discovered that there is a great deal of unexpected flexibility in this binding region, indicating that instead of only one there might be multiple ways that inhibitors could bind to the integrase enzyme.
The most recently reported of these simulations is in an article by Julie Schames, Richard Henchman, Jay Siegel, Christoph Sotriffer, Haihong Ni, and McCammon in the Journal of Medicinal Chemistry of April 8, 2004. These simulations revealed that a "trench" opens intermittently in the area of the integrase catalytic site, suggesting that it should be possible to develop inhibitors that bind with their cyclic moieties to the "east" of the active center as well as to the "west." Based on these simulations, the researchers were also able to predict that the strongest binding inhibitors should be possible with moieties on both sides, what the scientists have called "butterfly" compounds with "wings" extending to both the east and west.
"This pointed the way to new classes of drugs," said McCammon. "And it's very exciting that scientists at Merck have now verified all of this experimentally."

Hope for Effective Treatments The simulations have pointed to new classes of drugs to treat HIV/AIDS. Models of two proposed "butterfly" inhibitors, which occupy both of the binding orientations found in the active site of the HIV-1 integrase. McCammon group, UCSD. |
Because of the great promise these compounds are showing for treating HIV/AIDS, the Merck scientists are proceeding with development of a series of drug candidates that rely on the remarkable flexibility revealed by the McCammon group's simulations in the integrase binding site. The first of these compounds is expected to go into clinical trials this year, with UCSD as one of the trial sites.
The Merck scientists have chosen a drug development strategy in which they are developing "single wing" compounds, whose wings, or cyclic moieties, can bind to either the west or the east in the integrase enzyme. "These compounds are especially interesting because they'll be less likely to lead to drug-resistant strains of HIV-1," said McCammon. This is because the inhibitor can flip to the east if an integrase mutation arises in the west, or to the west if a mutation arises in the east. And because emerging HIV-1 mutations have significantly reduced the usefulness of many previously effective drugs, maintaining the ability to bind even when mutations occur is an important advantage.
"These results, combining our computer simulations at SDSC and Merck experimental research, are giving real hope that the remarkable story of molecular dynamics simulations in helping discover HIV-1 protease inhibitors may be repeated with HIV-1 integrase inhibitors," said McCammon. "This could potentially save many lives in the fight against HIV/AIDS."
The molecular dynamics simulations, carried out at SDSC and other sites, and the development of advanced molecular dynamics computational methods in McCammon's group, were supported by the National Science Foundation. This research has also been supported by the National Institutes of Health, the Howard Hughes Medical Institute, the NSF Center for Theoretical Biological Physics, the National Biomedical Computation Resource, the W.M. Keck Foundation, and Accelrys, Inc.